レジメンの切り替えおよび簡素化の管理 MANAGING REGIMEN SWITCH &
SIMPLIFICATION
抗HIV薬による治療歴の多い患者に対する治療選択肢

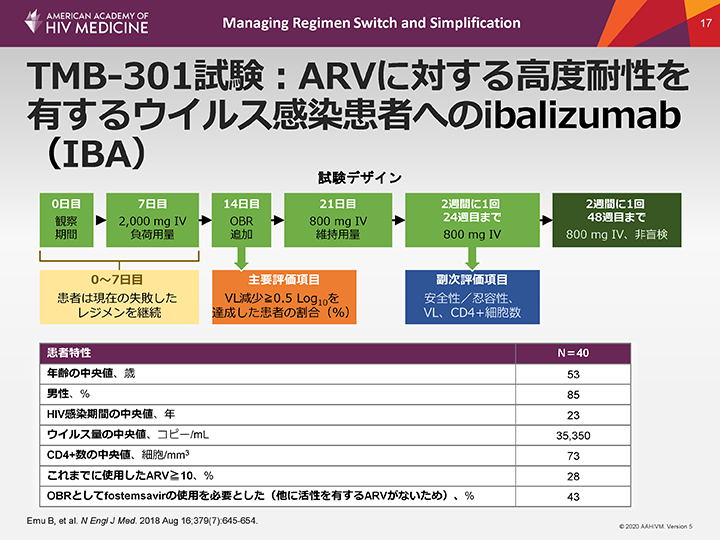
TMB-301試験:ARVに対する高度耐性を有するウイルス感染患者へのibalizumab(IBA)
Ibalizumab(IBA)は、post attachment阻害作用をもつ長時間作用型モノクローナル抗体である。この薬剤は、高度耐性HIVに感染しており、治療選択肢が限られている患者に対して開発され、試験対象となった患者集団が限られていたことから、FDAからオーファンドラッグとして承認された。
第III相試験であるTMB-301試験では、患者は最適化された併用薬の投与レジメン(OBR)とともに2週ごとにIBAの静脈内投与を受けた。治療レジメンが失敗しており、耐性検査で評価した3つのARVクラスそれぞれにおいて1剤以上に対する耐性を示し、ARVによる治療歴の多い患者40例をこの試験に組み入れた。OBRを構成するため、それぞれの患者はさらに1剤以上のARVに感受性がある必要があり、感受性がない場合は、当時治験薬であったfostemsavirの投与を受ける意思がある者とした。
この患者集団の年齢の中央値は53歳、85%が男性、HIV感染期間は20年(中央値 23[2-30] )以上であった。CD4+細胞数の中央値は73細胞/mm3であった。ウイルス量の中央値は35,350コピー/mLであった。患者の28%はこれまでに10種類以上のARVによる治療を受けており、また、約半数(43%)はfostemsavirをOBRに含める必要があった。
6日間の観察期間後、7日目に失敗したレジメンを継続しながら全例にIBAを2,000 mgの負荷用量で静脈内投与した(機能的単剤療法としてのIBA)。14日目にOBRを追加し、21日目にIBA 800 mgの静脈内投与を開始し、24週目まで隔週で投与した。最初の24週間を完了した患者は、拡大アクセスプログラム(TMB-311試験)の一環として、さらに24週間IBAの継続投与が可能であった。
患者27例が非盲検の拡大アクセスプログラムに組み入れられた。
ART:抗レトロウイルス療法、ARV:抗HIV薬、IBA:ibalizumab、IV:静脈内投与、OBR:最適化された併用薬の投与レジメン、VL:ウイルス量
IBAに関するTMB-301試験:結果

14日目において、患者の83%でウイルス量がベースラインから0.5 log10以上減少し、60%で1.0 log10以上減少した。24週目には、55%で1.0 log10以上の減少、48%で2.0 log10以上の減少が認められ、平均減少量は1.6 log10であった。この時点で、43%がウイルス学的抑制(50コピー/mL未満)を達成し、半数は200コピー/mL未満であった。
24週目の全体のCD4+細胞数の平均増加量は、約50細胞/mm3であった。CD4+細胞数の増加量は48週目まで維持された。
拡大アクセス試験に組み入れられた27例では、ウイルス量減少の中央値は24週目で2.5 log10であり、これは48週目まで維持された。24週目にHIV-RNA量が検出限界値未満(<50コピー/mL)であった15例全例が、48週目においてもウイルス学的抑制を維持していた。さらに1例が48週目までにこの閾値を下回った。
この試験で認められた有害事象のほとんどは軽度または中等度であった。1例は24週目に免疫再構築症候群(IRIS)発症のため治療を中止し、4例はその他の理由により中止し、4例は高度の免疫抑制により死亡した。
IBAは、米国で治療歴の多い患者に対して承認された(2018年3月)。
IBA+OBRの投与を受けた多剤耐性HIV-1感染患者38例を対象とした別の拡大アクセスコホート試験であるTMB-311試験により、7日目に76%の患者が0.5 log10以上のVL減少を達成した。24週目の来院を完了した患者24例のうち、46%がウイルス量50コピー/mL未満であり、48週目の評価を受けた17例のうち、47%がウイルス量50コピー/mL未満を達成した。7例は市販のIBA投与を受けるために試験を中止したため、48週目の評価項目が得られなかった。いずれの時点でも、VLのベースラインからの変化量の中央値は -2.6 log10コピー/mLであった。安全性および忍容性の結果はTMB-301試験と同様であった。
IBA:ibalizumab
BRIGHTE試験:治療歴の多い成人患者に対するfostemsavir96週間投与
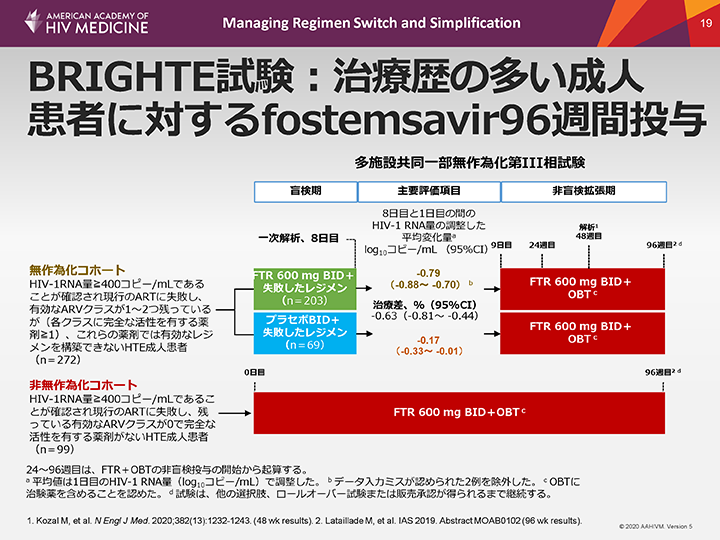
治療歴の多い(HTE)患者に対するARTを評価したもう1つの試験が、BRIGHTE試験である。この試験では、治療歴の多い患者にfostemsavir(FTR)+至適基礎療法(OBT)投与を実施した。
FTRは、治療歴の多いHIV-1感染患者のために開発されたファースト・イン・クラスの接着阻害剤プロドラッグである。8日間の機能的単剤療法後に、プラセボよりも優れた有効性が示された(主要評価項目)。FTR投与群では、HIV-1 RNA量が約0.8 log10コピー/mL減少したのに対し、プラセボ群では0.17 log10コピー/mL減少した(P<0.001)。
48週目には、無作為化コホート54%と非無作為化コホート38%の患者にウイルス学的抑制(HIV-1 RNA量40コピー/mL未満)が認められた。無作為化コホート60%(272例中163例)と非無作為化コホート37%の患者が、96週目までHIV-1 RNA量40コピー/mL未満を維持した。非無作為化コホート81%の患者において、FTRのみが完全な活性を有するARVであった。FTRの忍容性は概ね良好であり、有害事象による中止例はほとんどみられなかった。
ART:抗レトロウイルス療法、ARV:抗HIV薬、BID:1日2回、FTR:fostemsavir、HTE:治療歴の多い、OBT:至適基礎療法
BRIGHTE試験:96週目の結果
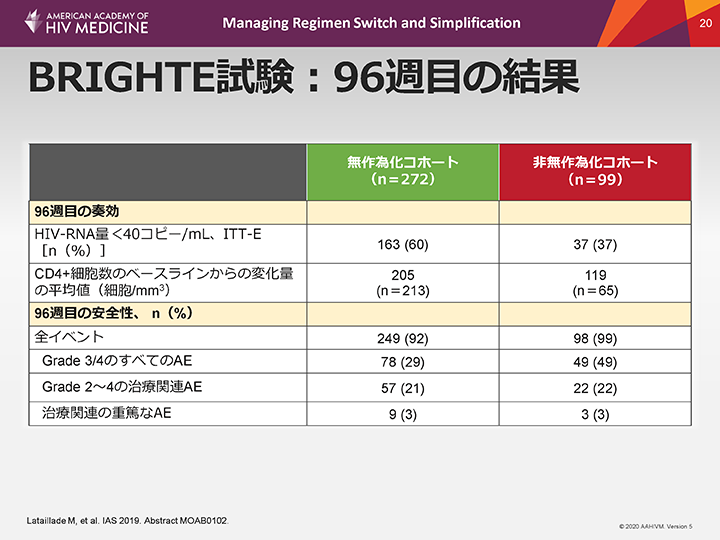
96週目には、無作為化コホート60%および非無作為化コホート37%の患者がウイルス学的抑制(ウイルス量40コピー/mL未満と定義)を達成した。
CD4+細胞数(細胞/mm3)の平均値は増加し、無作為化コホートでは205細胞/mm3増加、非無作為化コホートでは119細胞/mm3増加した。
奏効率は、女性、黒人、高齢患者で高かったが、ベースラインのHIV-1 RNA量が100,000コピー/mL以上またはベースラインのCD4+細胞数が20細胞/mm3未満の患者ではより低かった。
FTRを含むレジメンの96週目までの忍容性は概ね良好であった。Grade 3/4の有害事象の発現率は、非無作為化コホートの方が高かった。治療関連有害事象として最も多かったのは、悪心、下痢、頭痛であった。
AE:有害事象、BL:ベースライン